Das Zellweger-Syndrom: Alles Wissenswerte
Das Zellweger-Syndrom ist eine Erkrankung, die die Funktionen wie den Muskeltonus oder die visuelle und auditive Wahrnehmung beeinträchtigt. Darüber hinaus betrifft sie auch das Gewebe der Knochen oder der Organe, wie das Herz und die Leber.
Das Zellweger-Syndrom wurde 1964 beschrieben und wird mit dem Vorhandensein bestimmter Genmutationen in Verbindung gebracht. Diese Mutationen werden durch die autosomal rezessive Vererbung übertragen. Mit anderen Worten, sowohl der Vater als auch die Mutter müssen die Mutationen in ihrem genetischen Material haben.
Kinder, bei denen das Zellweger-Syndrom diagnostiziert wurde, neigen dazu, zu sterben, bevor sie ein Jahr alt werden. Viele sterben jedoch bereits vor dem sechsten Lebensmonat.
Der Tod tritt in der Regel als Folge von Veränderungen der Leber, der Atemwege und des Magen-Darm-Systems ein. Menschen mit milden Varianten dieser Erkrankung können jedoch bis ins Erwachsenenalter leben.
Das Zellweger-Syndrom

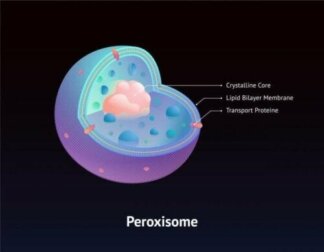
Das Zellweger-Syndrom gehört zu einer Gruppe von Krankheiten, die dieselbe genetische Ursache haben. Diese Ursache sind Störungen der Peroxisom-Biogenese, auch Zellweger-Spektrum-Störungen genannt. Peroxisomen sind Organellen, die bei der ordnungsgemäßen Funktion einiger Enzyme eine Rolle spielen.
Derzeit wissen Experten, dass es sich dabei nicht um unabhängige Veränderungen handelt. Es hat sich gezeigt, dass es sich dabei um unterschiedliche Schweregrade von Störungen der Peroxisom-Biogenese handelt.
Das klassische Zellweger-Syndrom ist die schwerwiegendste Variante von Peroxisomen-Biogenese-Störungen. Fälle mittlerer Schwere werden jedoch als neonatale Adrenoleukodystrophie bezeichnet. Die milderen Fälle sind in dieser Hinsicht als infantile Refsum-Krankheit bekannt.
Das könnte dich auch interessieren: Wissenswertes über genetische Mutationen
Charakteristische Symptome und Anzeichen
Beim Zellweger-Syndrom kommt es durch eine Veränderung der Biogenese des Peroxisoms zu neurologischen Ausfällen. Aus diesem Grund gibt es eine Vielzahl von Symptomen, die verschiedene Körpersysteme und -funktionen betreffen.
Im Folgenden findest du die häufigsten und charakteristischsten Symptome des Zellweger-Syndroms:
- Verminderter Muskeltonus
- Augen- und Sehstörungen
- Schwierigkeiten bei der Nahrungsaufnahme
- Beeinträchtigung der normalen körperlichen Entwicklung
- Vorhandensein charakteristischer Gesichtszüge wie ein abgeflachtes Gesicht, eine hohe Stirn und eine breite Nase
- Ebenso das Vorhandensein anderer morphologischer Veränderungen und Anomalien der Knochenstrukturen
- Erhöhtes Risiko für die Entwicklung von Herz-, Leber- und Nierenerkrankungen
- Atemwegserkrankungen wie Apnoe
- Erhöhte Lebergröße und Auftreten von Zysten darin
- Erkennung von Anomalien bei der elektroenzephalographischen Aufzeichnung und Anfälle
- Schließlich allgemeine Veränderungen in der Funktion des Nervensystems
Das Zellweger-Syndrom und seine Ursachen

Das Zellweger-Syndrom wurde mit dem Vorhandensein von Mutationen in mindestens 12 Genen in Verbindung gebracht. Obwohl es Veränderungen in mehreren Genen geben kann, ist ein verändertes Gen bereits ausreichend.
In etwa 70 % der Fälle befindet sich die Mutation im PEX1-Gen. Wie wir bereits erwähnt haben, wird die Erkrankung durch einen autosomal rezessiven Vererbungsmechanismus übertragen.
Das bedeutet, dass eine Person eine mutierte Kopie des Gens von jedem ihrer Eltern erben muss, um die typischen Symptome des Zellweger-Syndroms aufzuzeigen. Wenn beide Elternteile das mutierte Gen tragen, besteht ein Risiko von 25%, an der Krankheit zu erkranken. Diese Gene stehen mit der Synthese und der Funktion von Peroxisomen in Verbindung.
Peroxisomen sind Teile der zellulären Struktur von Organen wie der Leber. Dieses Organ übt äußerst wichtige Funktionen aus, wie zum Beispiel die folgenden:
- Fettsäurestoffwechsel
- Entsorgung von Abfallstoffen
- Allgemeine Gehirnentwicklung
Unser Lesetipp für dich: Welche äußerlichen Eigenschaften können Kinder genetisch erben?
Behandlung und Umgang mit der Krankheit
Derzeit ist keine Behandlung bekannt, die die tief greifenden Anomalien, die das Zellweger-Syndrom verursacht, behebt. Daher ist die Behandlung dieser Krankheit symptomatisch.
Fütterungsprobleme sind ein relevantes Symptom, da sie aufgrund der Schwierigkeit, Lebensmittel zu schlucken, das Risiko einer Mangelernährung bergen. Daher kann das Kind eine Magensonde benötigen, um Entwicklungsstörungen zu minimieren.
Die Behandlung dieses Syndroms ist multidisziplinär. In diesem Sinne können die Behandlungsteams aus Fachleuten der Pädiatrie, Neurologie, Orthopädie, Augenheilkunde und Chirurgie bestehen.
Alle zitierten Quellen wurden von unserem Team gründlich geprüft, um deren Qualität, Verlässlichkeit, Aktualität und Gültigkeit zu gewährleisten. Die Bibliographie dieses Artikels wurde als zuverlässig und akademisch oder wissenschaftlich präzise angesehen.
- Li, X., Baumgart, E., Morrell, J. C., Jimenez-Sanchez, G., Valle, D., & Gould, S. J. (2002). PEX11 beta deficiency is lethal and impairs neuronal migration but does not abrogate peroxisome function. Molecular and Cellular Biology.
- Klouwer, F. C. C., Berendse, K., Ferdinandusse, S., Wanders, R. J. A., Engelen, M., & Poll-The, B. T. (2015). Zellweger spectrum disorders: clinical overview and management approach. Orphanet Journal of Rare Diseases. https://doi.org/10.1186/s13023-015-0368-9
- South, S. T., & Gould, S. J. (1999). Peroxisome synthesis in the absence of preexisting peroxisomes. Journal of Cell Biology. https://doi.org/10.1083/jcb.144.2.255
Dieser Text dient nur zu Informationszwecken und ersetzt nicht die Beratung durch einen Fachmann. Bei Zweifeln konsultieren Sie Ihren Spezialisten.







