Ewing-Sarkom: Symptome und Behandlungen dieser seltenen Krebsart


Beim Ewing-Sarkom bilden sich Knochentumore, die sich durch Klumpen, Schwellungen, Knochenschwäche, Steifheit, Gewichtsverlust, Müdigkeit und sogar spontane Brüche bemerkbar machen.
Obwohl Risikofaktoren wie das Alter oder die ethnische Zugehörigkeit festgestellt wurden, sind die genauen Ursachen für das Ewing-Sarkom noch nicht bekannt. Zu den Behandlungen gehören unter anderem Strahlen- und Chemotherapie.
Die Überlebensrate bei dieser Krebsart liegt zwischen 60 und 70 %.
Was ist ein Ewing-Sarkom?
Das Ewing-Sarkom ist eine seltene Krebsart, die die Knochen oder das umliegende Weichteilgewebe angreift und Tumore bildet. Diese können sich auf andere Bereiche ausbreiten und Metastasen bilden.
Bei Erwachsenen ist es nicht häufig. Im Allgemeinen sind eher Kinder und Jugendliche betroffen, meist Jungen. Das ist vor allem während der Pubertät der Fall, wenn die Knochen im vollen Wachstum sind. Die Inzidenz wird auf 2,73 Fälle pro Million Menschen geschätzt.
Obwohl sie ähnliche Erscheinungsformen haben können, gibt es Unterschiede zwischen Osteosarkom und Ewing-Sarkom. Was die Inzidenz angeht, so tritt das erstere häufiger auf. Sie entstehen aus unterschiedlichen Zellen.
Dieser Artikel könnte dich ebenfalls interessieren: Arten von Sarkomen und deren Merkmale
Ewing-Sarkom: Anzeichen und Symptome
Das Ewing-Sarkom tritt häufiger in den Knochen der unteren Gliedmaßen, insbesondere im Oberschenkelknochen, sowie im Becken auf. Es kann aber auch an anderen Stellen auftreten.
Zu den Anzeichen und Symptomen gehören die folgenden:
- Knochenschmerzen: Sie können stoßweise auftreten und nachts weniger stark sein.
- Schwellung: Gefühl von Klumpen, die sich weich anfühlen können.
- Empfindlichkeit im betroffenen Bereich.
- Steifheit in der Nähe des Gelenks.
- Schwäche oder Taubheit in den Gliedmaßen.
- Spontane Knochenbrüche.
- Knochentumore
Einige dieser Anzeichen können auf andere Erkrankungen zurückzuführen sein. Es ist jedoch wichtig, dass du einen Arzt/eine Ärztin aufsuchst, wenn du sie bemerkst. Allerdings gibt es auch Menschen mit einem Ewing-Sarkom, die keine Symptome haben.
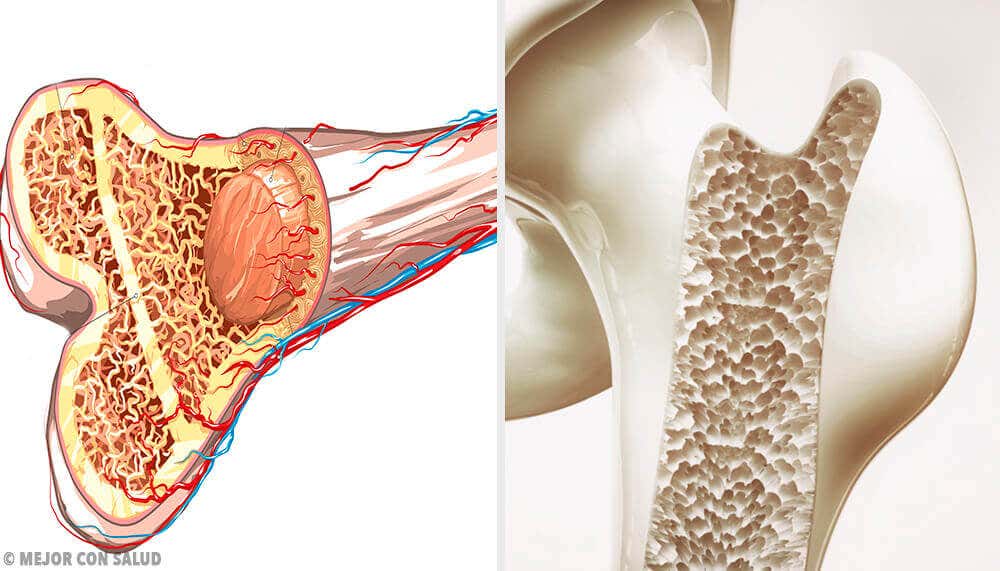
Ursachen und Risikofaktoren
Die genauen Ursachen des Ewing-Sarkoms sind noch nicht geklärt. Es ist bekannt, dass diese Krankheit beginnt, wenn Zellen Veränderungen in ihrer DNA erfahren, die dazu führen, dass sie sich unkontrolliert vermehren.
Diese Veränderungen betreffen am häufigsten ein Gen, das als EWSR1 bekannt ist. Dadurch entsteht eine Tumormasse aus Zellen, die sich ausbreiten und in andere Knochen metastasieren kann.
Darüber hinaus ist bekannt, dass diese Krankheit nicht vererbbar ist. Zu den Risikofaktoren für das Ewing-Sarkom gehören die folgenden:
- Alter: Obwohl dieses Sarkom in jedem Alter auftreten kann, treten die meisten Fälle bei Menschen zwischen 10 und 20 Jahren auf.
- Geschlecht: Die Krankheit tritt häufiger bei Männern auf.
- Ethnie: Menschen mit schwarzer Hautfarbe oder asiatischer Herkunft sind seltener betroffen als Menschen europäischer Herkunft.
Lies auch diesen Artikel: Morbus Paget: Erkrankung der Knochen
Wie erfolgt die Diagnose?
Die Diagnose des Ewing-Sarkoms beginnt mit einer körperlichen Untersuchung. Außerdem befragt der Arzt/die Ärztin den Patienten/die Patientin oder die Eltern nach den Symptomen, der Häufigkeit und der Dauer der Beschwerden. Auf dieser Grundlage lassen sich verschiedene Tests anordnen.
Bildgebende Tests
Bildgebende Tests beginnen mit Röntgenaufnahmen, bei denen offensichtliche Tumormassen entdeckt werden können. Außerdem erfolgen CT-Scans, vor allem im Brustbereich, um festzustellen, ob sich der Tumor auf die Lunge ausgebreitet hat.
Auch MRT-Scans können zu diesem Zweck zum Einsatz kommen. Schließlich werden noch eine Positronen-Emissions-Tomographie und eine Knochenszintigraphie durchgeführt.
Biopsien
Es wird eine Nadelbiopsie durchgeführt. Dabei wird eine Nadel durch die Haut in den Tumor eingeführt, um eine Probe zu entnehmen. Anschließend erfolgt eine labortechnische Untersuchung der Probe.
Alternativ kann eine chirurgische Biopsie nach einer Operation zur Entfernung des Tumors durchgeführt werden. Es gibt aber auch die Möglichkeit, nur einen Teil des Gewebes durch einen kleinen Einschnitt zu entfernen.
Weitere Tests
Zusätzlich zu den oben genannten Untersuchungen erfolgen verschiedene Labortests:
- Vollständiges Blutbild
- DNA-Tests, um festzustellen, ob es genetische Mutationen gibt
- Andere Tests, mit denen sich erhöhte Laktatdehydrogenase (LDH)-Werte feststellen lassen.
Behandlungsoptionen für das Ewing-Sarkom
Anhand des Krankheitsverlaufs, des Zustands des Patienten und seines Alters entscheidet das Ärzteteam über das weitere Vorgehen. Im Folgenden beschreiben wir die gängigsten Behandlungsmethoden beim Ewing-Sarkom.
Chemotherapie
Die Behandlung beginnt in der Regel mit einer Chemotherapie. Dabei werden leistungsstarke Medikamente kombiniert, um die Krebszellen abzutöten.
Die Medikamente können oral oder intravenös verabreicht werden. Zu den gebräuchlichsten gehören Cyclophosphamide, Doxorubicin, Vincristin und Dactinomycin.
Wenn der Tumor noch nicht gestreut hat, wird die Chemotherapie alle 2 Wochen verabreicht. Auch wenn eine Operation erforderlich ist, kann die Behandlung mit diesen Medikamenten nach der Operation fortgesetzt werden.
Die Chemotherapie hat oft Nebenwirkungen wie Haarausfall, Appetitlosigkeit, Übelkeit, Erbrechen, Müdigkeit und ein erhöhtes Infektionsrisiko durch die Entwicklung einer Neutropenie.
Chirurgischer Eingriff
Bei einer Operation geht es darum, die Krebszellen vollständig zu beseitigen. Die kompliziertesten Fälle gehen bis hin zur Amputation einer Gliedmaße.
In anderen Fällen lässt sich der Tumor ohne Amputation entfernen. Da jedoch ein Teil des Knochengewebes verloren geht, ist die Funktion der Gliedmaße beeinträchtigt.
Es können Transplantate erforderlich sein, entweder aus dem eigenen Knochengewebe des Patienten oder von einem Spender, sowie Implantate (Metall- oder Kunststoffprothesen).
Strahlentherapie
Die Strahlentherapie ist ebenfalls eine Option nach der Operation, um verbleibende Krebszellen zu zerstören. Im Allgemeinen hilft sie, das Krebswachstum zu reduzieren und Symptome wie Schmerzen zu lindern.
Zu den Nebenwirkungen gehören Müdigkeit, Hautreaktionen und Magenbeschwerden. Langfristig kann die Behandlung das Knochenwachstum beeinträchtigen und sogar das Risiko einer erneuten Krebserkrankung erhöhen.
Andere Verfahren
Neben den oben genannten gibt es weitere Verfahren zur Behandlung des Ewing-Sarkoms. Dazu gehört die Knochenmarktransplantation, entweder allogen oder autolog.
Sie kann in Kombination mit einer Chemo- oder Strahlentherapie erfolgen. Ihre Wirksamkeit wird noch erforscht.
Weitere Betreuung für Patienten mit einem Ewing-Sarkom
Auch nach Abschluss der Behandlung sollten Patienten mit einem Ewing-Sarkom weiter untersucht und überwacht werden. In diesem Zusammenhang werden die folgenden Empfehlungen ausgesprochen:
- Durchführung von Röntgenaufnahmen im Abstand von sechs Monaten über einen Zeitraum von fünf Jahren. Danach sollten sie für den Rest des Lebens jährlich erfolgen.
- Außerdem sollte ein regelmäßiger Lungen- und Knochen-Scan erfolgen.
- Darüber hinaus muss der Patient nach der Operation an einer Physiotherapie teilnehmen, um die Beweglichkeit wiederzuerlangen oder zu lernen, eine Gliedmaße zu benutzen, falls Implantate vorhanden sind.
- Es gibt auch emotionale Unterstützungsdienste, die bei einem Verlust von Gliedmaßen helfen.
Ewing-Sarkom: Prognose und Komplikationen
Gelegentlich spricht der Körper nicht auf die Behandlung an, und das Sarkom kann nicht kontrolliert werden, was zu einer fortgeschrittenen oder tödlichen Erkrankung führt. In der Regel sind die Verfahren jedoch in einer hohen Zahl von Fällen erfolgreich.
Die Zahlen variieren. Die 5-Jahres-Überlebensrate wird für Kinder unter 15 Jahren auf 75 % geschätzt. Dieser Prozentsatz nimmt mit zunehmendem Alter leicht ab und liegt bei Jugendlichen bei 58 %. Das Gleiche gilt, wenn Metastasen vorhanden sind.
Es kann zu einer dauerhaften Remission oder nur zu einer vorübergehenden Remission kommen, sodass der Krebs zurückkehrt. In den ersten 2 Jahren nach Beendigung der Behandlung ist die Wahrscheinlichkeit eines Rezidivs größer.
Spätrezidive (bis zu 5 Jahre später) sind beim Ewing-Sarkom häufiger als bei anderen Krebsarten. Bei einem Wiederauftreten kann die Behandlung schwieriger sein, da die Medikamente, die für die Chemotherapie verwendet wurden, wegen des Risikos der Toxizität nicht erneut verschrieben werden können.
Alle zitierten Quellen wurden von unserem Team gründlich geprüft, um deren Qualität, Verlässlichkeit, Aktualität und Gültigkeit zu gewährleisten. Die Bibliographie dieses Artikels wurde als zuverlässig und akademisch oder wissenschaftlich präzise angesehen.
- Hernández González E, Mosquera Betancourt G, Quintero Martínez O, Hernández Cabezas I. Sarcoma de Ewing. AMC. 2013; 17(5): 623-640.
- Jiménez Soto D. Sarcoma de Ewing en Pelvis. Revisión bibliográfica. San José: Universidad de Costa Rica, 2019.
- Khanna N, Pandey A, Bajpai J. Metastatic ewing’s sarcoma: Revisiting the “Evidence on the Fence”. Indian J Med Paediatr Oncol. 2017; 38: 173-181.
- Sánchez J. Lactato deshidrogenasa. Bol. Hosp. San Juan de Dios. 1999; 46(3): 182-187.
- Muñoz Villa A. Tumores óseos. Rabdomiosarcomas. Pediatr Integral 2016; XX (7): 458 – 464.
- Thowinson-Hernández M, Hernández-Martínez A. Neutropenia febril inducida por quimioterapia e infecciones asociadas: una revisión de la literatura. Gaceta Mexicana de Oncologia. 2019; 18: 1-6.
- Horowitz M, Kinsella T, Wexler L, et al. Total-body irradiation and autologous bone marrow transplant in the treatment of high-risk Ewing’s sarcoma and rhabdomyosarcoma.. Journal of Clinical Oncology. 1993; 11(10):1911-1918.